英茂盛业生物科技有限公司
023-67630383
023-67630383
 销售咨询QQ2548969917 技术咨询QQ1291769782
销售咨询QQ2548969917 技术咨询QQ1291769782热线/微信:13681365274
销售咨询QQ2548969917 技术咨询QQ1291769782微信扫一扫
下载说明书


实验内容
293T-tet-spCas9细胞是稳转四环素诱导的spCas9的细胞株。我们针对TP53基因设计的靶点构建到pLVshRNA载体,包装为慢病毒后,转染293T-tet-spCas9细胞。诱导spCas9后,实现对TP53基因敲除。
1、本实验基因敲除原理:293T-tet-spCas9细胞产生spCas9,与pLVshRNA载体产生的gRNA结合,,gRNA将诱导Cas9蛋白对靶点DNA进行切割,造成DNA断裂。细胞通过邻端连接( nonhomologous end-joining)修复重新连接DNA,造成靶点附近DNA序列缺失突变。
2、pTYNE载体靶点验证原理:将包括靶点在内的大约200bp基因组序列克隆到pTYNE载体。构建好的pTYNE载体与pCas9/gRNA1基因敲除载体共转染293T细胞。如果靶点有效,基因敲除载体对pTYNE载体载体切割,经过细胞修复后pTYNE上移码突变的EGFP基因得到修复,发出绿色荧光。
简要步骤
设计靶点→构建pCas9/gRNA1载体→pTYNE验证靶点→构建靶点表达慢病毒载体→稳转293T-tet-spCas9细胞→诱导spCas9表达→测序验证基因敲除
靶点设计和基因敲除载体构建
人TP53基因有多个mRNA和蛋白拷贝,2个转录起始位点,以及多个翻译位点。我们在TP53基因的第四外显子设计了2个CRISPR基因敲除靶点,以实现对所有TP53蛋白的编辑。
靶点序列1:acctgccctgtgcagctgt
靶点序列2:ttgattccacacccccgcc
将靶点1和靶点2构建到pCas9/gRNA1载体,获得针对TP53基因的gRNA基因敲除载体pCas9/gRNA1-P531和pCas9/gRNA1-P532。构建方法同pCas9/gRNA1使用说明中的方法。
靶点验证
1、pTYNE-53验证载体构建
为了验证基因敲除靶点的效率,我们把TP53第四外显子部分序列通过XhoI、HindIII酶切位点连接到pTYNE载体,通过pTYNE载体验证靶点的切割效率。构建完成的载体命名为pTYNE-53。
构建完成后pTYNE-53载体序列:

注: 黄色:CMV promoter;灰色:TP53第四外显子部分序列;绿色:EGFP序列;下划线:CRISPR靶点
2、用pTYNE-53验证靶点
1) 转染前24小时将293T铺到24孔板中。同时准备3个孔。
2) 按照下列体系制备2个质粒稀释液
组1(阴性对照) | 组2(靶点一) | 组3(靶点二) |
pTYNE-53 0.4ug | pTYNE-53 0.4ug | pTYNE-53 0.4ug |
pCas9/gRNA1空载体0.4ug | pCas9/gRNA1-P531 0.4ug | pCas9/gRNA1-P532 0.4ug |
DMEM培养基X ul | DMEM培养基 X ul | DMEM培养基 X ul |
总体积50ul | 总体积50ul | 总体积50ul |
3) 准备转染试剂稀释液:
Polyfect-V转染试剂 4.8ul
DMEM培养基 145.2ul
4) 将质粒稀释液和转染试剂稀释液分别混匀。取50ul转染试剂稀释液分别加入两组质粒稀释液中,充分混匀,室温孵育15min。
5) 将转染液加入293T细胞。
6) 48小时后检测EGFP荧光表达。
验证实验结果
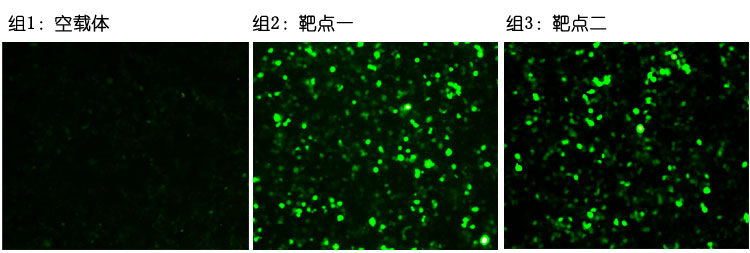
图一:靶点效率检测。转染48小时候荧光照片。
检测结果显示靶点一和靶点二均有效果,靶点一的效果优于靶点二。
TP53基因敲除293T细胞筛选
实验步骤
1) 将靶点一构建到pLVshRNA载体,并包装为慢病毒,稳转293T-tet-spCas9细胞,得到293T-tet-spCas9-TP53gRNA细胞。
2) 在293T-tet-spCas9-TP53gRNA细胞加入DOX 100ng/ml诱导spCas9 48小时。
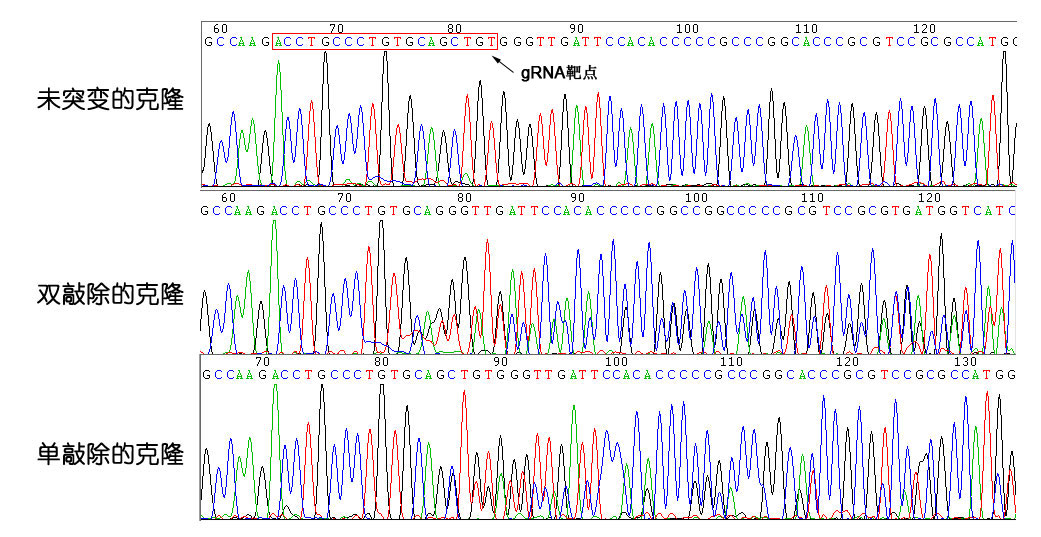
3) 分选细胞克隆,采用基因组PCR测序检测目的基因编辑情况。
克隆检测结果
以细胞克隆提取出的基因组DNA为模板,PCR扩增TP53第4外显子基因序列,并测序。
测序的20个克隆结果如下:
单敲除细胞克隆 | 12个 |
双敲除细胞克隆 | 2个 |
未突变细胞克隆 | 6个 |
合计 | 20个 |
测序结果截图: